RBM.py
Reciprocal Best Matching Algorithm
Introduction
Attempts to annotate unknown DNA sequences using a traditional bioinformatics approach - reciprocal best matching - but with further basing in statistics and integer programming.
Features
- Based on statistics, information theory, graph theory, and integer programming so you know at least some effort went into it
Download
| RBM.zip | RBM.tar.gz | View on GitHub |
Dependencies
Demonstration
I previously designed a Python script that annotates contigs for my bioinformatical research, but I felt that I took the easy way and ignored indels completely. Looking at other papers, I see that others attempt to endow sequences in strange fashions as well, such as arbitrary yet blind significance cutoffs, titling based off of the number of reads tiling, and assigning names with sequences that reciprocally "hit" each other using BLAST. Pragmatic from a scripting sense, but not fully justifiable from a theoretical perspective. Thus, I attempted to endeavour to persevere in creating a mathematically based algorithm for annotating contigs.
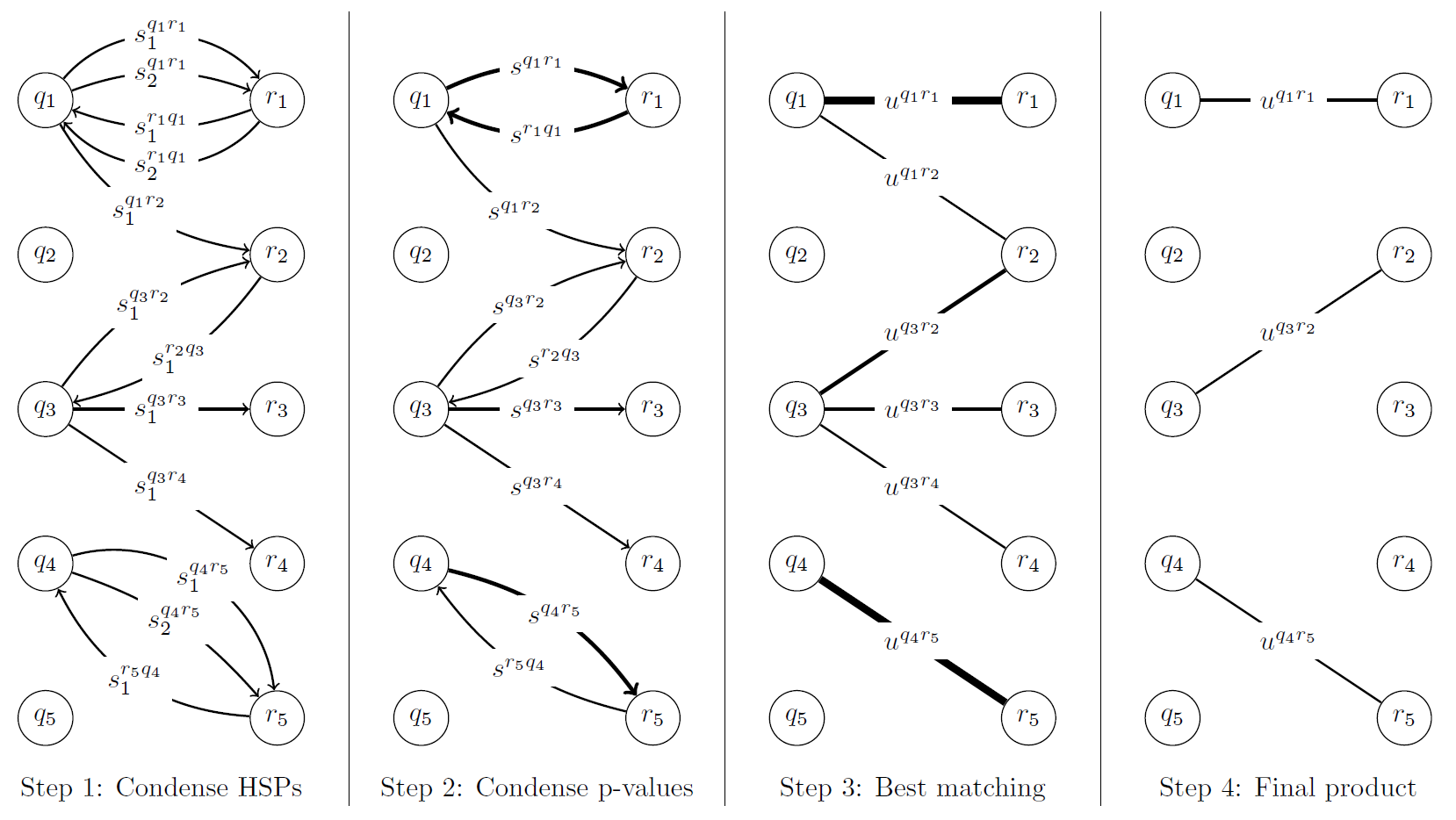
I will now detail how the reciprocal best matching algorithm is conducted. Beforehand, there exists two sets of sequences: the queries or unknown sequences and the references, which identities are known. The preliminary step is to BLAST the set of queries to the references, then in a reciprocal manner, the references to the queries. As a reminder, BLAST attempt to match each query to a reference and if significant (i.e. above a certain user defined threshold) will assign a "hit" between these two. There may be one or several high-scoring segment pairs (HSPs) between these two sequences, each one portraying their sequence similarity and as well, assigning a tangible score for each HSP. With the setup done, the first step is to condense these HSP scores into a single similarity score, which is accomplished using the properties of the Poisson distribution. Secondly, these reciprocal scores (which happen to be p-values) are merged into a single value utilizing some techniques found in information theory. Finally, a maximal matching is performed via integer programming.
- ls
- Queries.fasta QueryToRef.xml References.fasta RefToQuery.xml
- python GetRBMData.py QueryToRef.xml RefToQuery.xml Matches.csv
- python ReverseComplementQueries.py Queries.fasta QueryToRef.xml Matches.csv
- python CheckRBMAlgo.py \
- -t 3 \
- -o identities.csv \
- Queries_ReverseComplement.fasta \
- References.fasta \
- Matches.csv
To run, use the commands seen above, ensuring that you have both sets of sequences as FASTA and XML files generated from BLASTing. The final output file, in this case would be identities.csv, holds the annotation of the unknown query sequences to the known references. To learn more about the algorithm I described above, I have written a short document outlining my inner thoughts, as well as a curt teaching of basic biology for those not well versed in the subject matter (this problem was given to my mathematical friends, hence the biology tutorial), found here: RBM Problem. I would like to personally thank Daniel Anania for his contribution in integer programming.